用AI炸场“生命元宇宙”,Meta蛋白质大模型深度解析
编辑按:本文转载至微信公众号“智东西”,飞鲸投研经授发布 。
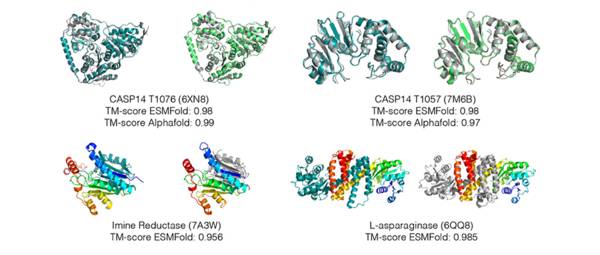
就在前几天,迄今为止参数最多、规模最大的蛋白质预测模型ESMFold被Meta官宣了,甚至有研究者宣称该模型又大又好,足以碾压Google在2021年推出的AlphaFold2。
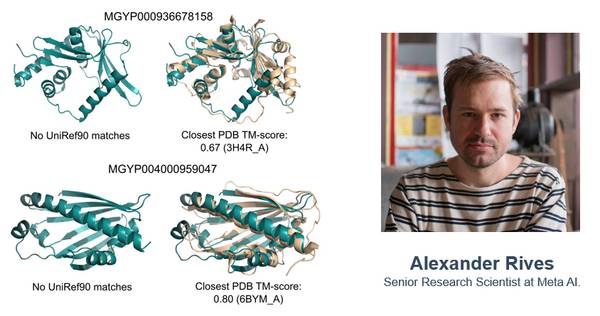
ESMFold与通讯作者Meta AI的Alexander
这一消息着实让学术界和工业界震撼,要知道这些大的模型,无论训练还是使用,都得有妥妥的“钞能力”,如果模型越来越小,说不定就不需要更大算力的芯片了。(当然事实并非如此)甚至LeCun大牛都发推为ESMFold背书,称之为“Super-fast and accurate”。
从氨基酸序列预测蛋白质结构是自然科学中长期存在的重大挑战。在基于进化的算法中,AlphaFold2可以说是目前解决该问题最成功的。它通过在多序列输入、进化同源物对齐序列和可选结构模板上训练端到端神经网络,取得了突破性成就,大大加速了“生命元宇宙”的构建。
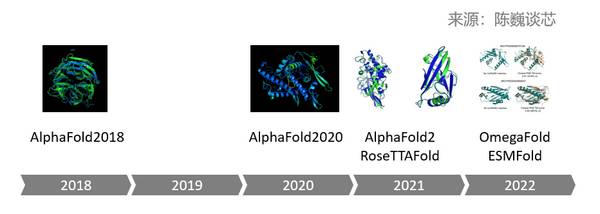
蛋白质预测AI大模型的进化
而Meta团队的ESMFold蛋白质模型只需要一个序列作为输入,该模型背后的团队由Meta AI(原Facebook AI)的资深研究科学家Alexander Rives主导。该团队专注于大规模蛋白质序列和结构数据的无监督表示学习模型研究。Alexander本人同时也是Fate Therapeutics、Syros Pharma、Kallyope的联合创始人,妥妥的科创家。
那ESMFold真的能碾压AlphaFold2吗?让我们先来回顾下什么是蛋白质结构预测,然后再深入分析ESMFold的网络结构。
ESMFold预测的结构
论文链接:https://doi.org/10.1101/2022.07.20.500902
01 什么是蛋白质结构预测?
首先,蛋白质结构是指各种蛋白质分子的空间结构。由线性氨基酸组成的蛋白质,需要折叠(Fold)成特定的空间结构,才具有相应的生理活性和生物学功能。
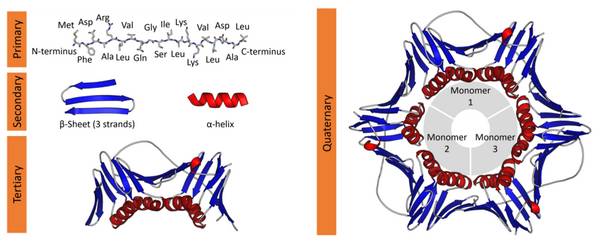
蛋白质的四级结构
蛋白质的分子结构可划分为四级,以描述其不同层级的特征:
蛋白质一级结构:组成蛋白质多肽链的线性氨基酸序列。
蛋白质二级结构:依靠不同氨基酸之间的C=O和N-H基团间的氢键形成的稳定结构,主要为α螺旋和β折叠。
蛋白质三级结构:通过多个二级结构元素在三维空间的排列所形成的一个蛋白质分子的三维结构。
蛋白质四级结构:用于描述由不同多肽链(亚基)间相互作用形成具有功能的蛋白质复合物分子。
我们所说的蛋白质结构预测(Protein Structure Prediction),就是指从蛋白质的氨基酸序列中预测蛋白质的三维结构。也就是说,从蛋白质的一级结构预测其折叠和二级、三级、四级结构。
DeepMind(Google旗下)的AlphaFold2在蛋白质结构预测大赛CASP 14中,对大部分蛋白质结构的预测与真实结构只差一个原子的宽度,达到接近冷冻电镜等复杂仪器检测的水平。这一巨大进步被Nature和Science选为2021年度十大科学突破。
根据不同的氨基酸和序列,蛋白质能折叠成的构型数量是一个天文数字,因此很难用常规方法进行蛋白质结构的准确预测。例如,目前实验的方法(例如冷冻电镜)至今才能解出10万的蛋白质结构。
冷冻电镜及其图像
因此,使用AI的方法,加速对蛋白质结构的解析,分析其组成和功能,就成了生物界和医药界的争相推进的重要工作。
02 ESMFold的“魔幻效果”
ESMFold与AlphaFold2和RoseTTAFold对多序列输入的蛋白质结构预测具有相当的准确度。但ESMFold突出优势在于,其计算速度比AlphaFold2快一个数量级,能够在更有效的时间尺度上探索蛋白质的结构空间。
过去,AlphaFold2和RoseTTAFold在原子分辨率蛋白质结构预测问题上取得了突破性成功,但依赖于使用多序列比对(Multiple Sequence Alignment,简写为MSA)和相似蛋白质结构的模板来实现最优表现。
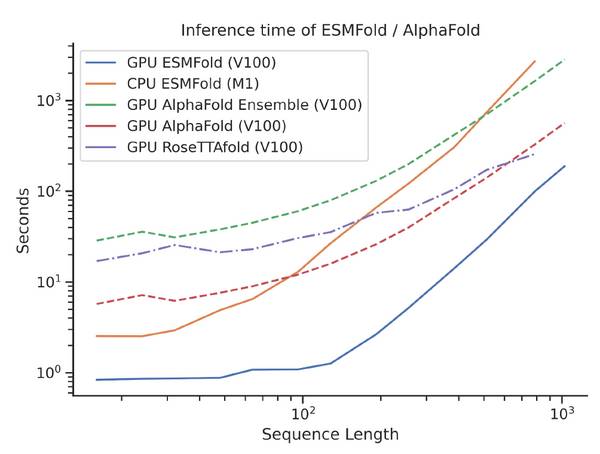
ESMFold模型具有比AlphaFold2更高的速度
ESMFold使用ESM-2学习的信息和表示来执行端到端的3D结构预测,特别是仅使用单个序列作为输入(AlphaFold2需要多序列输入),方便研究者在使用时通过模型缩放,将模型大小控制在数百万到数十亿量级参数。需要注意的是,随着模型大小的增加,可观察到预测准确性的持续提升。(还是“越大越准”)
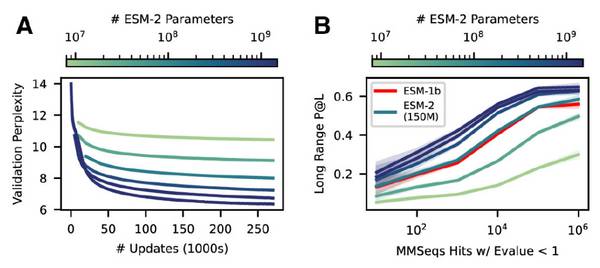
ESM-2模型随着参数量升高精度升高
由于ESMFold的预测速度比现有的其他原子分辨率蛋白质结构预测模型快一个数量级,因此ESMFold可以帮助快速构建蛋白质结构数据库。使用ESMFold,可以快速计算100万个预测结构,这些结构代表了蛋白质预测空间的不同子集,其中大多数没有注释的结构或功能。
而且ESMFold的大部分高置信度预测与已知的实验结构的相似度都很低,这表明了通过AI计算获得的基因组蛋白的结构新颖性。
值得注意的是,许多高置信度结构与UniRef90中的结构也具有低序列相似性,说明该模型具有超出其训练数据集的泛化能力,实现了基于结构的蛋白质功能预见能力。
据此,研究人员认为,ESMFold可以帮助理解那些超出现有认知的蛋白质结构。
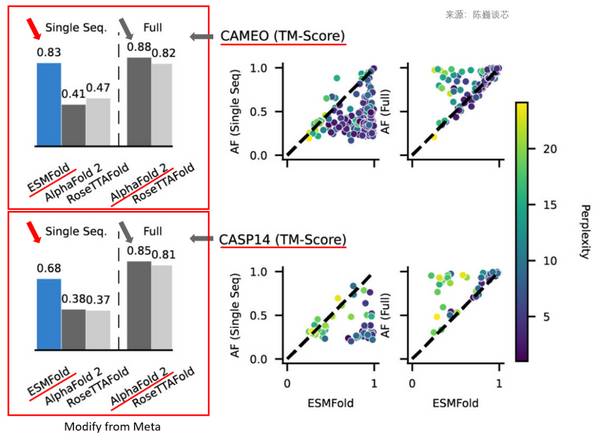
ESMFold在单序列输入时预测精度明显好于AlphaFold2
虽然ESMFold速度很高,精度也不错,特别是在单序列输入的时候精度明显好于AlphaFold2。但我们也要看到,ESMFold在多序列输入的情况下,其精度比AlphaFold2还是略有差距。
03 ESMFold网络结构
与AlphaFold2模型类似,ESMFold模型的架构也可以分为四部分:数据解析部分、编码器部分(Folding Trunk)、解码器部分(Structure Module)、循环部分(Recycling)。
ESMFold和AlphaFold2之间的一个关键区别是使用语言模型表示来消除对显式同源序列(以MSA的形式)作为输入的要求。
语言模型表示作为输入提供给ESMFold的折叠主干。通过将处理MSA的计算量大的Folding Block模块替换为处理序列的Tranformer模块来简化AlphaFold2中的Evoformer。这种简化或优化意味着ESMFold会比基于MSA的模型快得多。
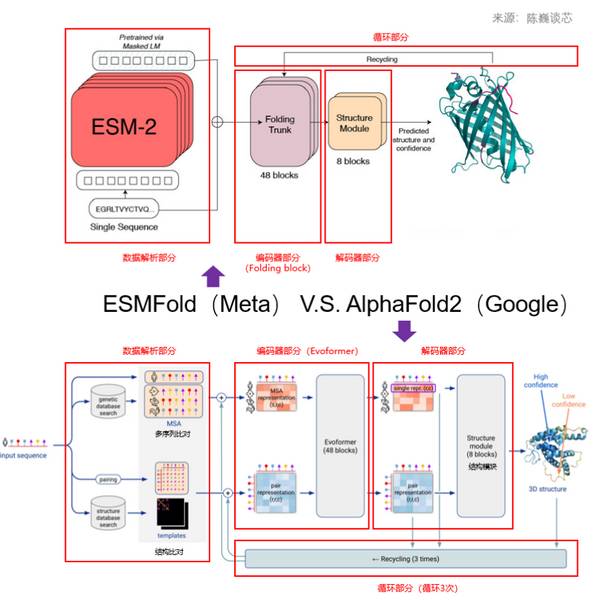
ESMFold与AlphaFold2对比
在AlphaFold2和RoseTTAFold中使用MSA和模板会导致两个瓶颈。
首先,可能需要基于CPU检索和对齐MSA和模板。这是由于AlphaFold2和RoseTTAFold不是二维序列嵌入状态,而是使用轴向注意力对应于MSA的三维内部状态进行操作,即使使用GPU,这一计算的代价也不菲。
相比之下,ESMFold是一个完全端到端的序列结构预测器,可以完全在GPU上运行,无需访问任何外部数据库。
例如在单个NVIDIA V100 GPU上,使用较少参数的ESMFold在14.2秒内对具有384个残基的蛋白质进行预测,可比单个AlphaFold2模型快6倍。而在较短的序列上,我们甚至看到了约60倍的改进。
速度的数量级提高是ESMFold优于AlphaFold2的独特优势,使我们能够在比现有方法更短的时间尺度内构建大量预测结构。考虑到可用序列数据的规模,这一点尤其重要。
例如,AlphaFold2蛋白质结构数据库的初始版本发布时具有约36万个预测结构,截至2022年7月则包含约99.5万个预测,这比目前许多蛋白质序列数据库小几个数量级。
04 数据解析部分与解码器的深度分析
数据解析部分用于输入序列和数据库的解析,为编码器提供输入。
在AlphaFold2模型中,数据解析部分使用了氨基酸序列数据库和结构数据库,分别用于相近序列的比对和结构模板的配对。

AlphaFold2多序列比对示意
生物信息学的基础是基于这样的一个假设:序列相似,结构相似,功能相似。一般认为相近的序列或者相近的结构会衍生出相近的功能域。
1)序列数据库被用于多序列比对(Multiple Sequence Alignment,MSA),即在序列数据库中检索与输入序列接近的数据库序列。
2)结构数据库则用于结构匹配,寻找与输入序列的结构接近的已知结构模板。
然后序列比对与结构比对的结果作为输入传输给编码器部分。
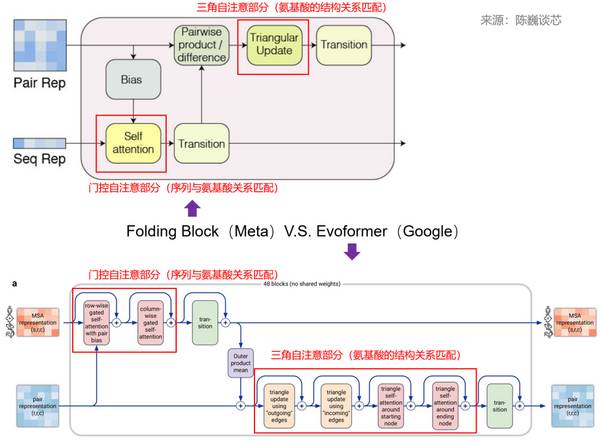
ESMFold Folding Block与AlphaFold2 Evoformer结构对比
解码器部分即Folding Trunk,一共48层。
ESMFold与AlphaFold2的一个关键区别是,ESMFold使用语言模型表示,消除了对明确的同源序列(以MSA的形式)作为输入的需要。
ESMFold通过用一个处理序列的Transformer模块取代处理MSA的计算昂贵的网络模块,简化了AlphaFold2中的Evoformer。这种简化意味着ESMFold的速度大大提高,远高于基于MSA的模型。
05 结语
作为蛋白质结构预测大模型,ESMFold获得准确原子分辨率结构预测的推断(Inferenc)速度比AlphaFold2提高了约一个数量级。特别是在实际计算中,这一速度优势表现的更加明显。这是由于ESMFold削减了搜索多序列来构建MSA的计算量。

ESMFold用于探索宏基因组结构空间
推断速度优势使得基于计算有效映射大型宏基因组序列数据库的结构空间成为可能。
除了用于识别远同源性外,ESMFold还可以被用于进行快速准确的结构预测,并在实际时间尺度内获得数百万个预测结构,进一步帮助发现新的蛋白质结构和功能。这相当于在使用AI计算来构建生命的“元宇宙”。
150亿参数大模型,10x倍速度提升。虽然Meta ESMFold精度上没能做到全面“碾压”AlphaFold2,但毕竟“唯快不破”,对于蛋白质结构解析与预测、构建大型宏基因组结构数据库有着巨大的推动作用。
参考文献:
Zeming Lin et. al., Language models of protein sequences at the scale of evolution enable accurate structure prediction, https://www.biorxiv.org/content/10.1101/2022.07.20.500902v1
Jumper, J. et al., Highly accurate protein structure prediction with AlphaFold, Nature (2021):1-11.
飞鲸投研从多维度分析,整理了一份《成长50》的名单,可以关注同名公众号:"飞鲸投研":feijingtouyan,进行领取(点击复制)
/阅读下一篇/
千亿SaaS巨头启示录,霸主之路还能走多久?



















脱水研报
-
中国邮政储蓄银行(PostalSavingsBankofChina)是中国一家大型零售商业银行,于2007年3月6日成立,总部位于北京市西城区。前身为邮政储金局
-
公司以陶瓷机械装备业务起家,并投资蓝科锂业布局锂电材料业务,实施双主业发展战略。科达制造股份有限公司前身为科达五金机械厂,于1992年建立,2002年改制为科达
-
公司成立于2007年4月,并于2021年7月在深交所创业板上市。主导产品为电力熔断器,下游主要为新能源汽车、风光发电及储能、通信、轨道交通等中高端市场,其中在国
-
2021年8月1日至9月13日,申万半导体指数涨跌幅为-17.13%,同期上证A指、沪深300指数、上证50指数和创业板指数涨跌幅分别为9.36%、3.75%、
-
新年新气象,物管行业喜获新政。1月5日,住房和城乡建设部等十部委发布《关于加强和改进住宅物业管理工作的通知》,加强和改进住宅物业管理工作。这标志着,物业管理行业





名家观点
-
一隐秀路大佬就是这波主多南天的主力,今天下午又再度拉回,从同花顺超级盘口看它从水下一路点火,要不是大盘太弱了大概率能走出地天,上次也是在一片绝望中隐秀路大佬引导
-
这一周的弱势,始于外资的大幅出逃,不过,周五的弱势,却怪不了人家,参考下北上资金,尽管深港通有一定流出,但也谈不上很大,沪港通更是流入的,所以,周五的弱,纯属于
-
以岭药业:这个票近期是一路小快步上行,到了今天终于是走了加速,明天溢价问题不大,但周四涨停也说明短线资金进来了,短期估计短线资金还会关注这里,明天预计冲高问题不
-
10月份已到了最后几天,三季报也进入到了最后的集中披露,而这个时候,就要注意下不及预期的雷股了。怎么规避不及预期的可能雷股?如果对个股基本面不是很了解,还真没什
-
第二段,就是11点之后,不管是中午前的强反击,还是午后的回落,以及随后的僵持,跟北上资金的节奏几乎完全同步了,这意味着,多空双方都选择了观望,然后,才有了北上资





热点题材
-
东吴证券认为,我国碳市场已具备总量控制和市场调控机制雏形,交易规模及覆盖行业存在较大提升空间。当前国内碳价低于海外,双碳目标时间紧任务重,总量收紧将驱动碳配额价
-
机构指出,光伏压延玻璃产能指标限制的放开,将在一定程度上降低行业扩产门槛,受益政策松绑扩产有望提速,但同时也解开了在资源、技术、资金、执行力等方面全面占优的行业
-
产业链调研显示,在气温走高及环保限产的影响下,上游企业停工检修情况较多,致使二氧化碳原料气供气不足,本周湖北、广东等地陆续有厂家进入检修期。当下正值二氧化碳行业
-
今年以来,L3级别自动驾驶能力的蔚来ET7、小鹏P5、华为极狐相继发布。在政策、造车新势力等多方势力共同推动下,自动驾驶正在加速落地。IDC预计到2024年,全
-
国泰君安分析师翟堃认为,一二级市场煤炭资产价格存在倒挂现象。鄂尔多斯近日公告,拟以49.8亿元收购永煤矿业(8.47亿吨可采储量)25%股权,吨可采储量对价23





最新资讯
-
而电力是数据中心的成本大头,2023年我国数据中心耗电量在全社会用电量中占比为3.3%。这些电力可以供14000个三峡水电站同时满负荷运转。在电力成本中,发电设
-
很多女生夏天开车为了防晒都要带上冰袖,为了解决这一痛点,小米su7采用了三层镀银前挡风玻璃,紫外线隔绝率达到了99.5%,红外线隔绝率97.6%,这也是目前轿车
-
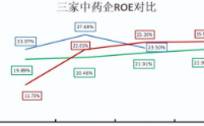
而成长赛道,注重盈利能力。在衡量公司盈利能力的指标中,ROE(净资产收益率:净利润/股东权益)最为全面。因为,ROE直接体现出公司利用股东权益赚取利润的能力。据
-
所以,基金经理的持仓变动,往往能给我们提供一个参考,同时也反映了资本偏爱的方向。知名基金经理张坤,在管4只基金,合计规模654.74亿元,其中规模最大的是易方达
-
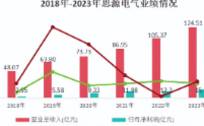
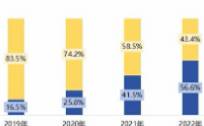
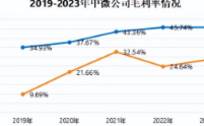
用这句话来形容中微公司在2023年的处境,是非常贴切的。在这一年里,半导体市场经历了21世纪以来最长的下跌期,内存和微处理器更是其中的“重灾区”。但同样是在这一